GNBF5040 final exam review
Genomics: Basic Concepts and Applications
| CUHK Student ID | Update |
|---|---|
| 1155228903 | 2024-12-02 |
1 Technology Platforms for Genomics
High-throughput Sequencing (HTS): Enables rapid sequencing of genomes and transcriptomes, providing comprehensive genetic information (1_CourseInfo_Introduction.pdf).
RNA Sequencing (RNA-Seq): Used to profile gene expression levels, identify novel transcripts, and discover post-transcriptional modifications (1_CourseInfo_Introduction.pdf).
2 Genomic Data Analysis
Quality Control: Essential for filtering and trimming low-quality reads to improve the accuracy of downstream analyses (3_SeuqncingDataAnalysis.pdf).
FASTQ File Inspection: Initially, the raw sequencing data in FASTQ format is inspected to assess the initial quality of the reads.
Running FASTP: FASTP is executed to perform quality control, which includes trimming low-quality ends, removing adapters, and filtering out reads that do not meet quality thresholds.
Quality Assessment with FASTQC: After preprocessing with FASTP, FASTQC is used to generate quality reports that visually display the quality of the sequences, confirming the effectiveness of the trimming and filtering step.
Genome Mapping: Aligns sequence reads to a reference genome to identify their genomic locations, which is crucial for variant detection and gene expression analysis (3_SeuqncingDataAnalysis.pdf).
Alignment to Reference Genome: Short sequences (reads) from sequencing are aligned to a reference genome to determine their positions within the genome.
Paired-End Information: For paired-end sequencing, the relative positions of the two reads are considered during alignment to improve accuracy.
Variant Detection: The results of genome mapping are often used for variant detection, including single nucleotide polymorphisms (SNPs) and insertions/deletions (Indels).
Structural Variation Identification: Genome mapping helps in identifying large structural variations such as inversions and translocations.
Coverage Analysis: Genome mapping reveals the sequencing coverage across different regions, which is crucial for assessing data quality and planning subsequent analyses.
Post-Processing: After mapping, BAM files need to be sorted, deduplicated, and indexed for efficient data retrieval and analysis.
3 Large Scale Genome Annotation Projects
Human Genome Project (HGP): An international research initiative aimed at mapping and sequencing the entire human genome, consisting of approximately 3 billion base pairs. Launched in 1990 and completed in 2003, it provided a comprehensive reference for human genetics, enabling advances in medicine, genetics, and biotechnology. (2_LargeGenomeProjects.pdf).
The Cancer Genome Atlas (TCGA): A comprehensive effort to map the genetic changes in various types of cancer, providing a valuable resource for understanding cancer biology and developing new therapies (2_LargeGenomeProjects.pdf).
4 Sequencing Data Analysis
Sequence Alignment: Identifies homologous sequences and functional elements across different genomes (3_SeuqncingDataAnalysis.pdf).
Input: Two or more sequences (query and reference or target sequences).
Scoring: Assign scores to matches, mismatches, and gaps based on a scoring system.
Alignment: Use algorithms (like Needleman-Wunsch, Smith-Waterman, or heuristic methods like BLAST) to find the optimal alignment that maximizes the score.
Output: The aligned sequences with gaps inserted to maximize similarity.
5 Genome Assembly
Genome Assembly: Reconstructs the full genomic sequence from short reads, which is fundamental in de novo genome sequencing projects (4_GenomeAssembly.pdf).

Input: Raw sequencing reads from an organism's genome.
Quality Control: Remove errors and low-quality data from the sequencing reads.
Read Trimming and Filtering: Trim adapter sequences and filter out reads that are too short or of poor quality.
Pre-assembly Read Correction: Correct errors in the reads to improve the accuracy of the assembly.
Overlapping: Determine overlaps between reads to create longer contiguous sequences called contigs.
Gap Filling: Identify gaps between contigs and attempt to fill them with additional sequencing data.
Scaffolding: Primarily, it uses the information from paired-end reads (read1 and read2) to determine the order and orientation of contigs. This helps in creating a more accurate and complete representation of the genome by aligning contigs based on the actual distances and sequences of the DNA fragments from which the reads were sequenced.

Contig Assembly: Assemble reads into longer sequences without gaps, known as contigs.
Scaffold Construction: Arrange contigs into scaffolds, which are sequences that represent larger genomic regions and maintain the order of contigs.
Gap Closure: Identify and close gaps within scaffolds to improve the continuity of the assembled genome.
Quality Assessment: Evaluate the quality of the assembly using metrics such as N50, which indicates the length of the smallest contig in a set of contigs that together contain at least 50% of the total assembly data.
Post-assembly Polishing: Refine the assembly by correcting errors and polishing the genome sequence.
Annotation: Identify genes, regulatory regions, and other functional elements within the assembled genome.
Output: A set of sequences representing the best approximation of the organism's genome based on the available sequencing data.
6 Genetic Variations
Point Mutation: A change in a single nucleotide that can alter gene function and is a source of genetic diversity (6_GeneticVariations.pdf).
SNP (Single Nucleotide Polymorphism): Common genetic variations used in genetic mapping and association studies (6_GeneticVariations.pdf).
Indel: Insertions or deletions of nucleotides that can disrupt gene structure and function (6_GeneticVariations.pdf).
CNV (Copy Number Variation): Variations in DNA copy number that can affect gene dosage and contribute to phenotypic variation (6_GeneticVariations.pdf).
Structural Variation (SV): Large-scale genomic rearrangements that can have significant effects on gene expression and disease susceptibility (6_GeneticVariations.pdf).
7 Transcriptional Regulation & ChIP-seq
ChIP-seq: Identifies the binding sites of transcription factors and histone modifications on a genome-wide scale, providing insights into gene regulation (7_TranscriptionlRegulation_ChIP-seq.pdf).
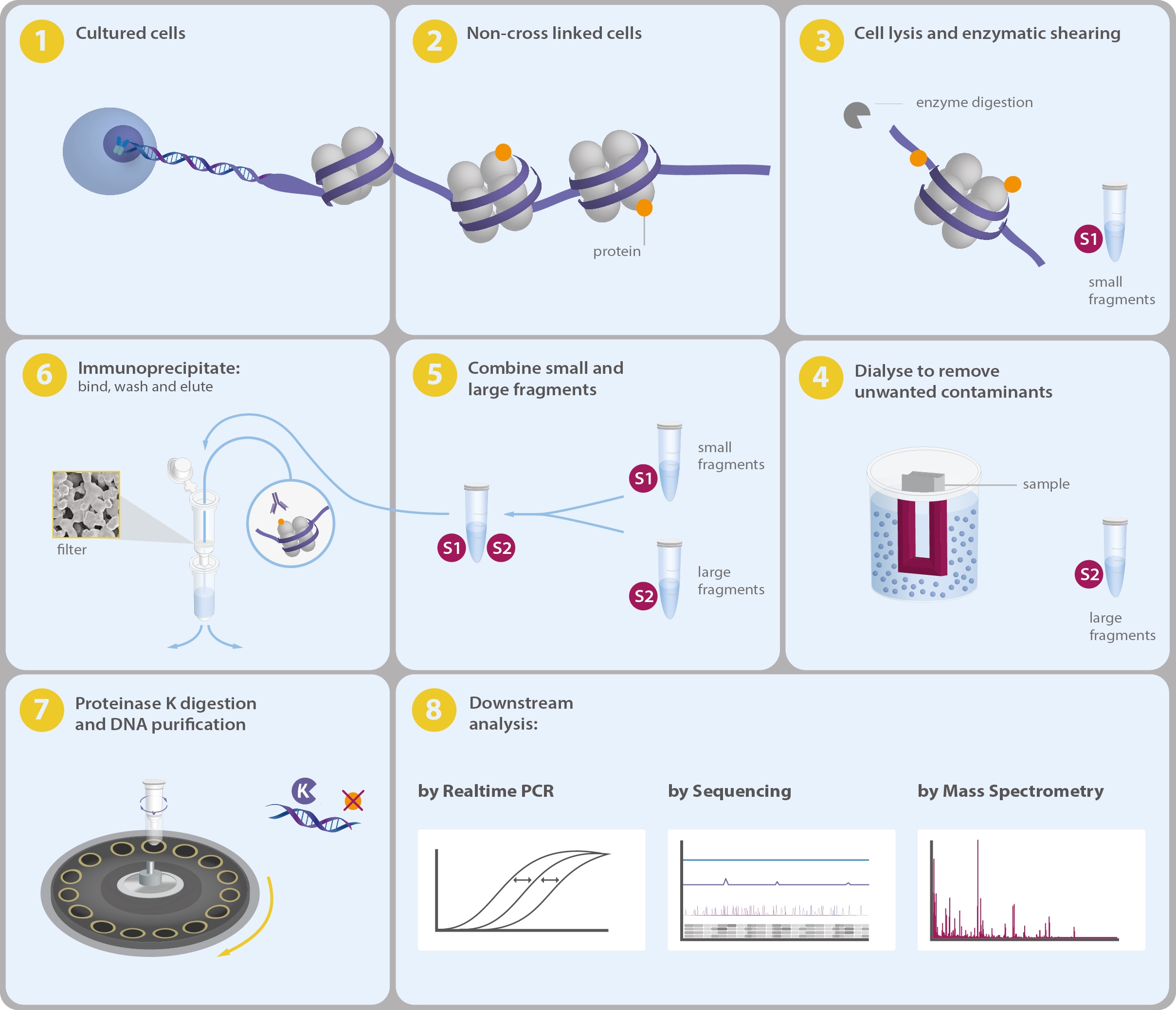
Crosslinking: Cells are treated with a crosslinking agent to fix protein-DNA interactions.
Fragmentation: Chromatin is broken into smaller pieces through sonication or enzymatic digestion.
Immunoprecipitation (IP): Use specific antibodies to precipitate protein-DNA complexes.
Reverse Crosslinking: Remove crosslinks to free DNA from proteins using heat or chemicals.
DNA Purification: Purify the DNA and prepare it for sequencing by repairing ends, adding adapters, etc.
Sequencing: High-throughput sequencing is performed to generate short DNA reads.
Bioinformatics Analysis:
Align reads to a reference genome to identify regions of enrichment.
Call peaks to determine specific DNA sequences bound by the protein.
Analyze data for motif enrichment and functional annotation of associated genes.
Validation and Interpretation: Validate results with additional experiments and interpret in the context of gene regulation.
Transcription Factors: Play a critical role in regulating gene expression by binding to specific DNA sequences (7_TranscriptionlRegulation_ChIP-seq.pdf).
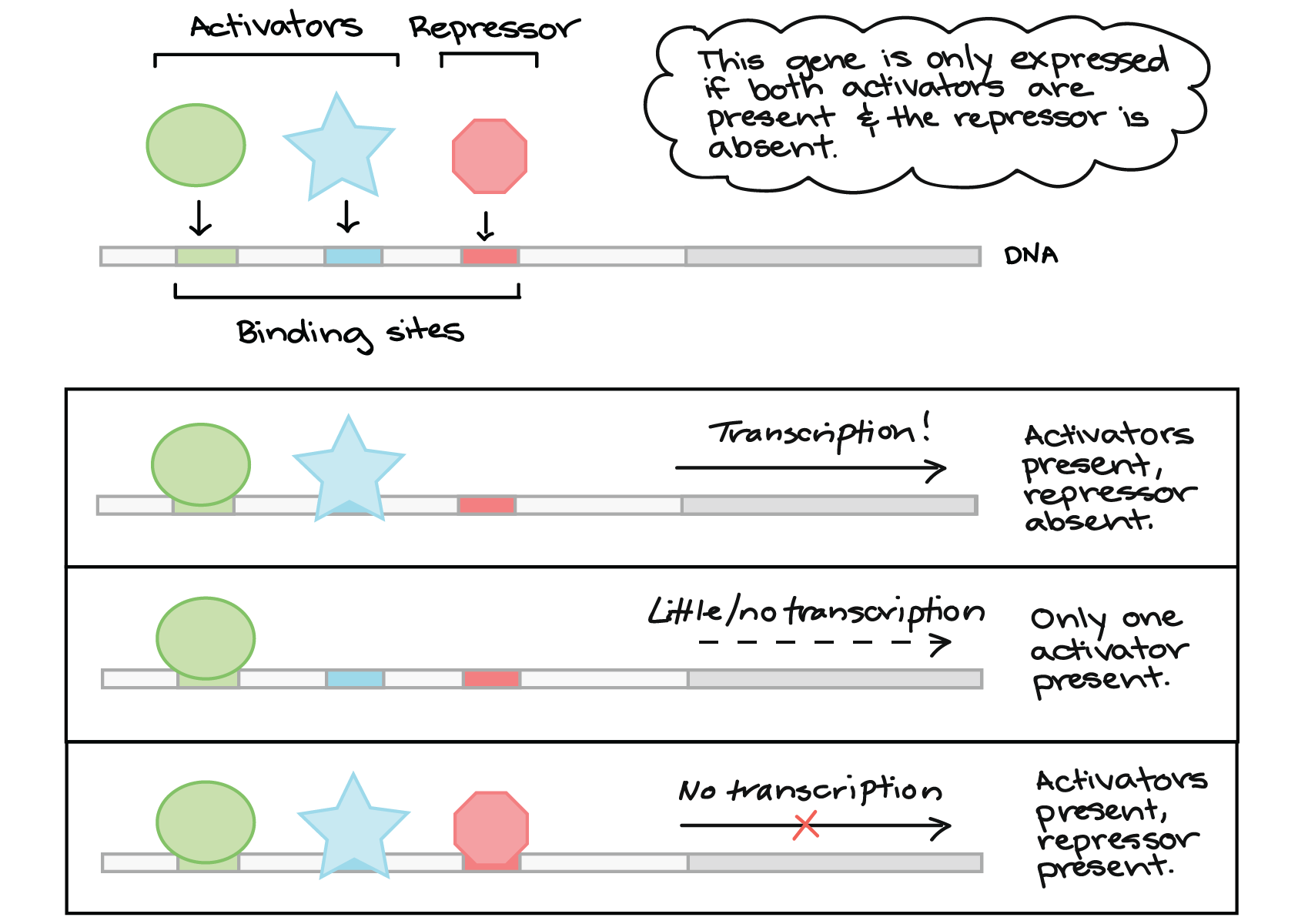
8 Epigenetics/Epigenome
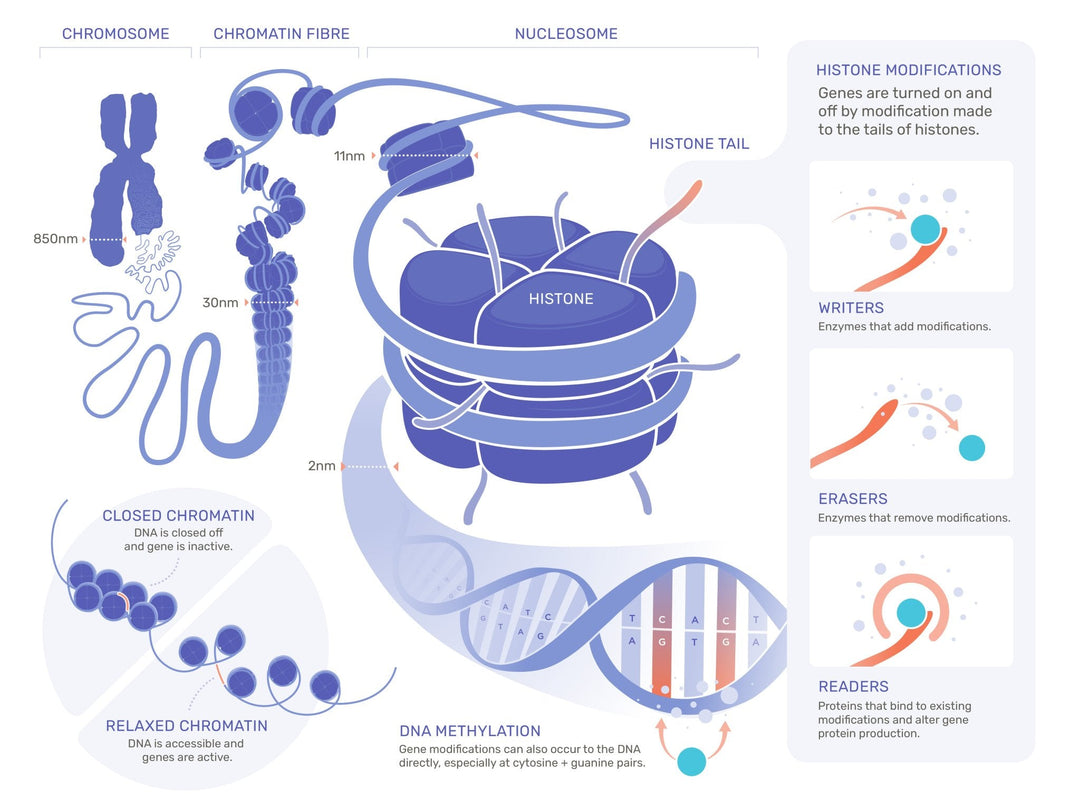
Epigenetics: Studies heritable changes in gene expression that do not involve changes to the underlying DNA sequence, such as DNA methylation and histone modifications (8_Epigenome_Epigenetics.pdf).
9 Transcriptome Sequencing / RNA-Seq
Transcriptome: Reflects the complete set of RNA transcripts produced by an organism under specific conditions, providing insights into gene expression dynamics (9_TranscriptomeSequecing.pdf).
Gene Expression Quantification: Normalized methods like FPKM account for sequencing depth and transcript length to accurately measure gene expression levels (9_TranscriptomeSequecing.pdf):
RNA-Seq workflow
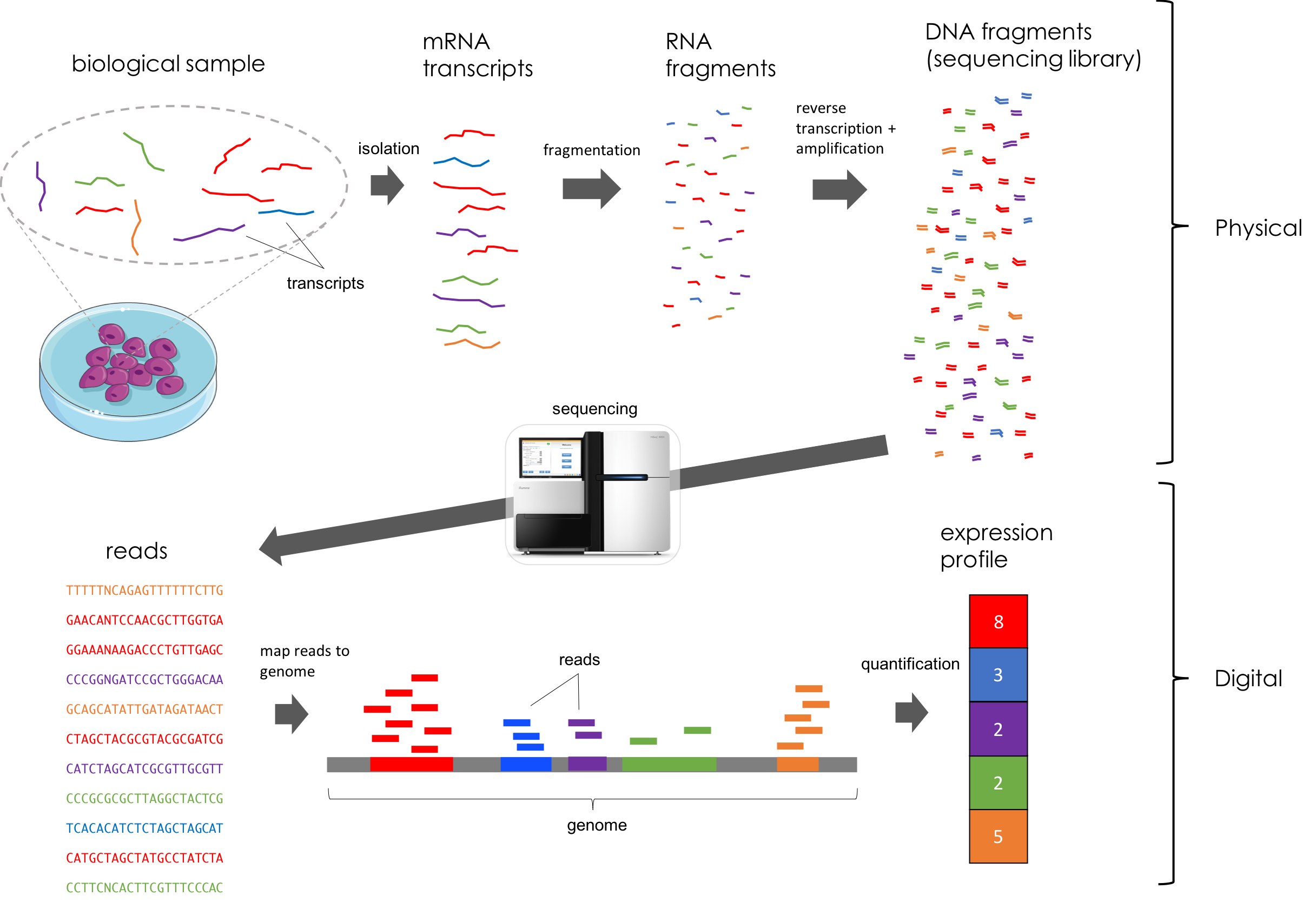
Sample Collection and RNA Extraction: Collect biological samples and extract total RNA.
RNA Quality Assessment: Evaluate the quality and quantity of the RNA to ensure it's suitable for sequencing.
Library Preparation: Prepare the RNA for sequencing by fragmenting and attaching adapters.
Sequencing: Generate short reads from the RNA molecules using high-throughput sequencing technology.
Read Alignment: Align the sequenced reads to a reference genome or transcriptome to identify corresponding genes and transcripts.
Quantification: Measure gene and transcript expression levels using metrics like RPKM or FPKM.
Differential Expression Analysis: Identify genes and transcripts with altered expression under different conditions.
Isoform Expression Analysis: Analyze alternative splicing and isoform expression patterns.
Functional Analysis: Conduct functional annotation and pathway analysis to interpret the biological significance of the expression changes.
Data Interpretation and Integration: Integrate RNA-Seq data with other data types for a comprehensive understanding of the biological system.
Validation: Validate the expression changes observed in the RNA-Seq data using independent methods like qPCR.
10 Single Cell Sequencing
Single Cell Sequencing: Reveals cellular heterogeneity by analyzing individual cells, allowing for the discovery of novel cell types and states (10_SingleCellSequending.pdf).
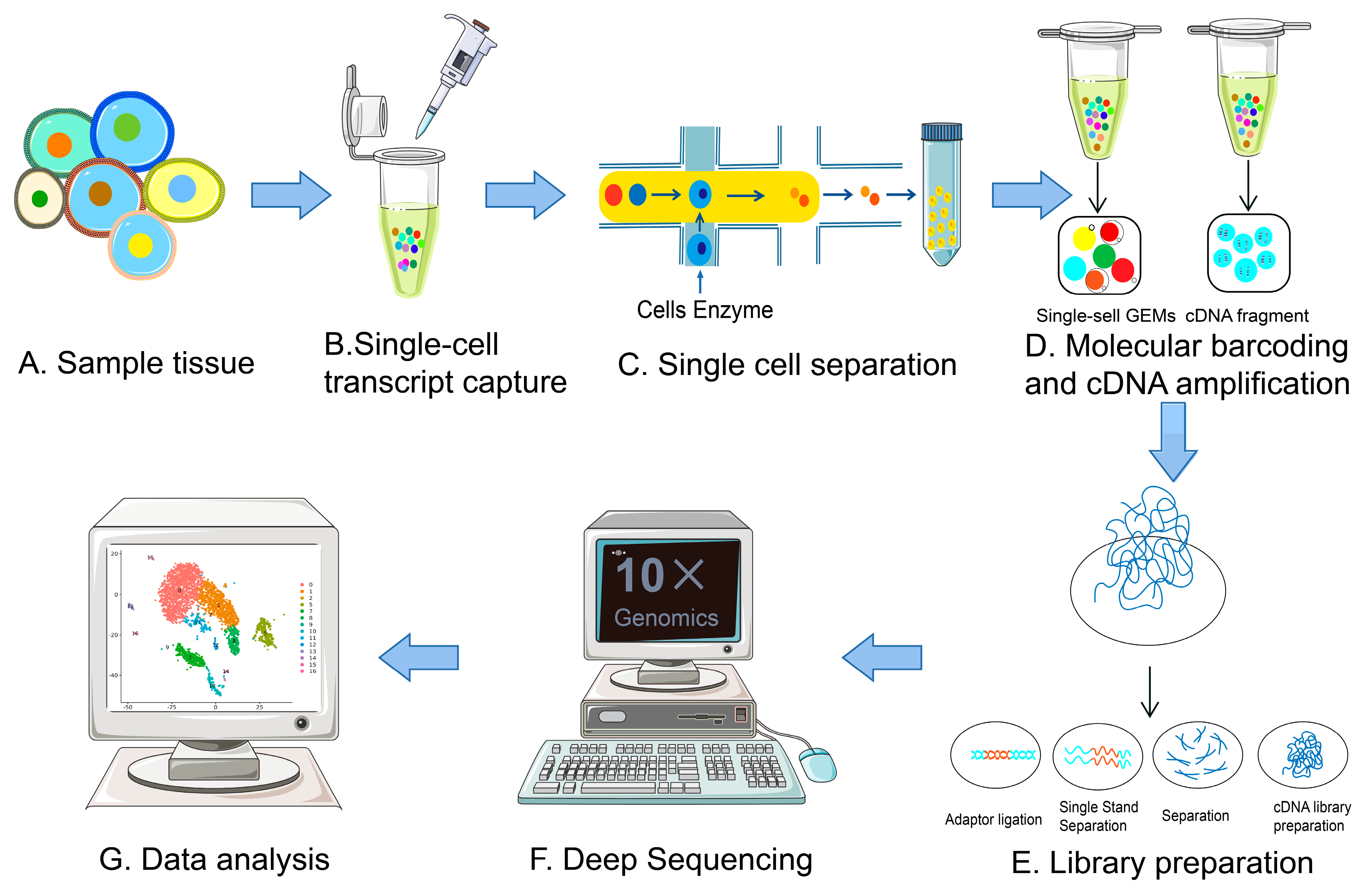
Cell Isolation: Individual cells are isolated from a tissue or population for separate analysis.
RNA Extraction: Extract RNA from each single cell, requiring specialized methods due to the small sample size.
Library Preparation: Prepare sequencing libraries from the RNA of individual cells, often incorporating unique molecular identifiers.
Sequencing: Sequence the libraries to generate reads that represent the transcriptome of each cell.
Read Alignment: Map the sequenced reads to a reference genome to determine the genes expressed in each cell.
Gene Expression Quantification: Quantify the expression levels of genes within each individual cell.
Data Analysis: Analyze the data to cluster cells based on transcriptomic profiles and identify distinct cell types or states.
Visualization: Use high-dimensional data visualization techniques like t-SNE or UMAP to represent cellular heterogeneity.
11 Non-Invasive Prenatal Testing (NIPT)
NIPT: Detects fetal chromosomal abnormalities by analyzing cell-free fetal DNA in maternal blood, offering a safer alternative to invasive diagnostic tests (11_NonInvasivePrenatalTesting.pdf).
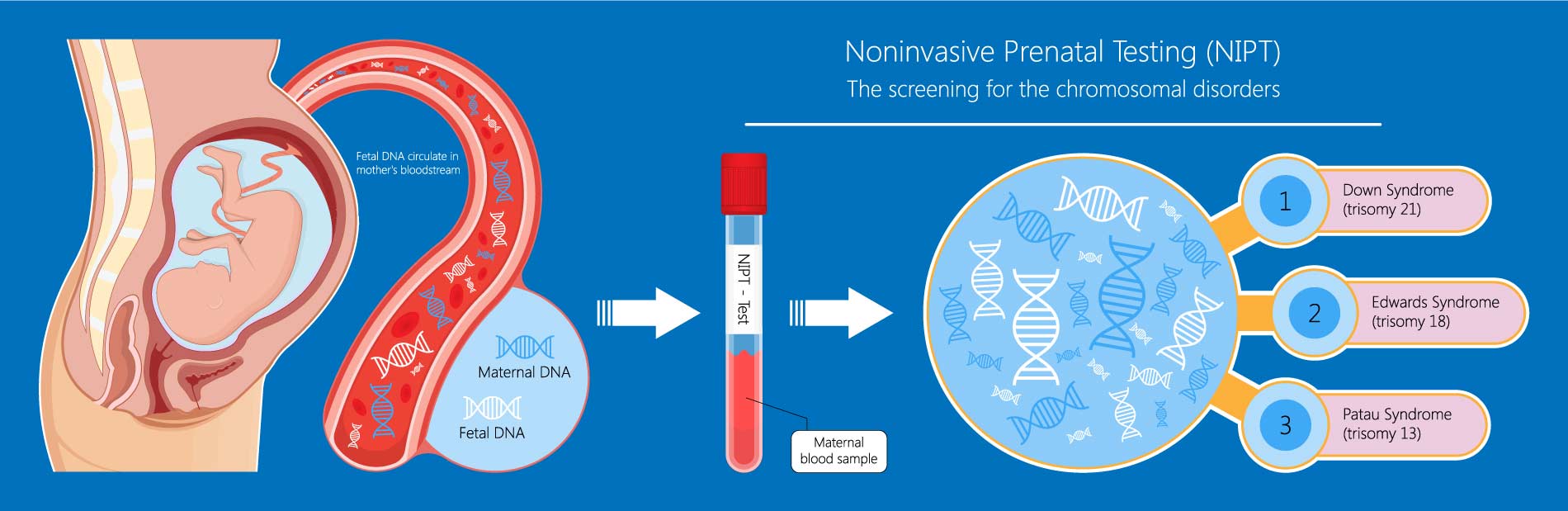
Genomic Representation (GR): A measure used in NIPT to determine the proportion of reads from a specific chromosome, aiding in the detection of aneuploidies (11_NonInvasivePrenatalTesting.pdf):